

선천성 당화장애
Congenital disorder of glycosylation
질병관리청
2020. 1. 1.
레어노트 등록일
2022. 6. 7.
조회
437
본 정보는 ‘질병관리청’에서 제공합니다.
질병관리청은 국민 보건 증진과 감염병 예방 및 관리를 위한 보건의료 기관입니다.
원인과 증상
원인
단백질의 당화과정에 필요한 효소의 유전적 결함
증상
발달 지연, 성장 부전, 간경변, 혈전증, 다기관 기능 부전
관련 부위
체내 : 간, 갑상선, 뇌, 생식기, 소장, 송과체, 위, 혈관(혈액)
체외 : 눈, 피부
진단과 치료
원인
혈청 transferrin의 등전점 전기영동분석, 유전자 검사
치료
보존적 치료 (mannose 경구 투여)
관련 질환
질환명
CDG syndrome
기타
산정 특례
질병코드
V900
의료비 지원
가능
개요
당화(glycosylation)는 대부분의 단백질과 지질 등이 기능을 하기 위해 거치는 대사과정으로 매우 광범위하고 복잡한 기전들이 관여하게 됩니다. 선천성 당화 장애(congenital disorder of glycosylation, CDG)는 당화과정에 필요한 효소의 부족으로 생기는 질환군을 통칭하는 질환으로, 유전 대사 이상 (inborn errors of metabolism)의 한 종류입니다. 임상증상은 원인에 따라 매우 다양한데, 신경학적 증상이 대부분을 보이며, 주로 심한 정신발달지체, 기형적인 신체발달, 소화기관의 문제로 인한 성장상재, 간기능 장애, 혈액응고 장애를 보이는 것으로 알려져 있습니다. 부족한 효소에 의해 당화작용의 장애가 어느 부위에서 일어나는지에 따라 대게 1형, 2형으로 나뉠 수 있으나 다양한 종류의 유전자 결함에 따라 세분화됩니다. 현재 100여종이 알려져 있으며, 가장 많은 아형(subtype)은 1a로 불렸던 phosphomannomutase-2 (PMM2) 결핍에 의한 PMM2-CDG이고 두 번째로는 glucosyltransferase I 결핍에 의한 ALG6-CDG입니다. CDG의 유병률은 유럽에서 10만 명당 0.1-0.5명으로 알려져 있으나, PMM2-CDG의 단일 아형으로만 보인자 정도가 2만-7.7만 명당 1명꼴이므로, 아직 미진단 환자가 많을 것으로 예상되며, 국내에도 검사의 제한으로 진단되지 않은 환자가 많을 것으로 추정되고 있습니다.
원인
인체 세포에는 단백질과 당질이 결합되어 생성된 다양한 종류의 당단백질(glycoprotein) 이 있습니다. 당단백질의 합성에는 단백질에 당질이 결합하는 당화(glycosylation) 가 일어나며 이 과정에는 여러 효소가 관여합니다. 이 효소의 유전적 결함으로 인하여 당화현상이 정상적으로 일어나지 않으면 인체에서 다양한 기능을 하는 당단백질이 만들어지지 않아 증상을 나타내게 됩니다. 당단백질은 인체의 다양한 조직에 분포하고 있기 때문에 다양한 기관에서의 증상을 나타낼 수 있으나 가장 대사가 활발한 기관인 신경계 및 근육 계통의 조직에서 가장 흔하게 증상이 나타납니다.
증상
일부 환자들의 경우 함몰 유두, 근 긴장 저하, 피하지방 침착 이상, 성기 모양 이상 등 출생 직후 확인이 가능한 증상을 가지고 태어납니다. 나머지 환자들은 생후 1세 이내에 정상적인 발달 과정이 나타나지 않는 발달지연이 나타나게 됩니다. 경련과 뇌졸중 유사 증상도 발달지연에 동반되어 흔하게 나타나며, 영상 검사를 할 경우 소뇌 부분의 위축이 흔하게 발견됩니다. 또한 키와 몸무게가 커 가는 성장 역시 또래에 비하여 저하되게 됩니다. 그 외에 간수치상승 및 간염, 색소망막염, 갑상샘 기능 저하증, 혈액 응고 장애, 피부 부종, 저혈당증, 면역력 저하 등이 함께 나타날 수 있습니다. 이러한 증상은 환자들의 유전자형에 따라서 다를 수 있으며 환자들에 따라 이러한 증상들 중 일부만 발현될 수 있습니다. 예후는 유전자형에 따라 매우 다양합니다. 약 20% 정도의 환자들은 생후 1년 내에 사망하며 그 외의 환자들도 정상 보행을 하기 어려운 경우가 많으나, 일부 환자의 경우에는 경미한 말초 신경 증상을 제외하고는 정상적인 생활을 할 수 있습니다.
진단
전반적인 성장 및 발달 지연을 보이는 환자에서 유전 대사 장애에 합당한 증상이 나타나는 경우에 의심할 수 있습니다. 채혈을 통해 얻어진 트랜스페린 (transferrin) 단백질의 당화 특성을 검사하여 선별합니다. 이러한 특성은 등점적 전기영동 (isoelectric focusing electrophoresis, IEF) 검사를 통해 분석합니다.
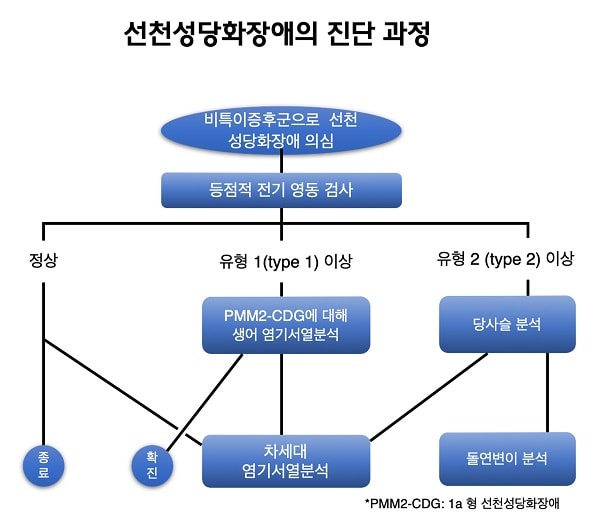
선천성 당화 장애가 의심되는 경우에는 유전자 검사를 통하여 확진하게 됩니다. 유전자 검사는 환자의 혈액을 채취하여 생어염기서열분석(Sanger sequencing)을 하거나 차세대 염기서열 분석법 (Next generation sequencing, NGS; Massive parallel sequencing)을 이용하여 검사할 수 있습니다. 차세대 염기서열 분석은 유전체의 염기서열을 고속으로 분석하는 방법을 말합니다. 기존의 생어 염기서열 분석과 달리 DNA를 수백만 개 이상의 조각으로 잘라서 각각의 염기서열을 분석한 뒤, 이렇게 얻는 데이터를 생물 정보학적 기법으로 합치게 됩니다. 이를 통해 방대한 유전체 정보를 빠르게 해독할 수 있게 하는 방법입니다.
선천성 당화 장애가 의심되는 경우에는 유전자 검사를 통하여 확진하게 됩니다. 유전자 검사는 환자의 혈액을 채취하여 생어염기서열분석(Sanger sequencing)을 하거나 차세대 염기서열 분석법 (Next generation sequencing, NGS; Massive parallel sequencing)을 이용하여 검사할 수 있습니다. 차세대 염기서열 분석은 유전체의 염기서열을 고속으로 분석하는 방법을 말합니다. 기존의 생어 염기서열 분석과 달리 DNA를 수백만 개 이상의 조각으로 잘라서 각각의 염기서열을 분석한 뒤, 이렇게 얻는 데이터를 생물 정보학적 기법으로 합치게 됩니다. 이를 통해 방대한 유전체 정보를 빠르게 해독할 수 있게 하는 방법입니다.
치료
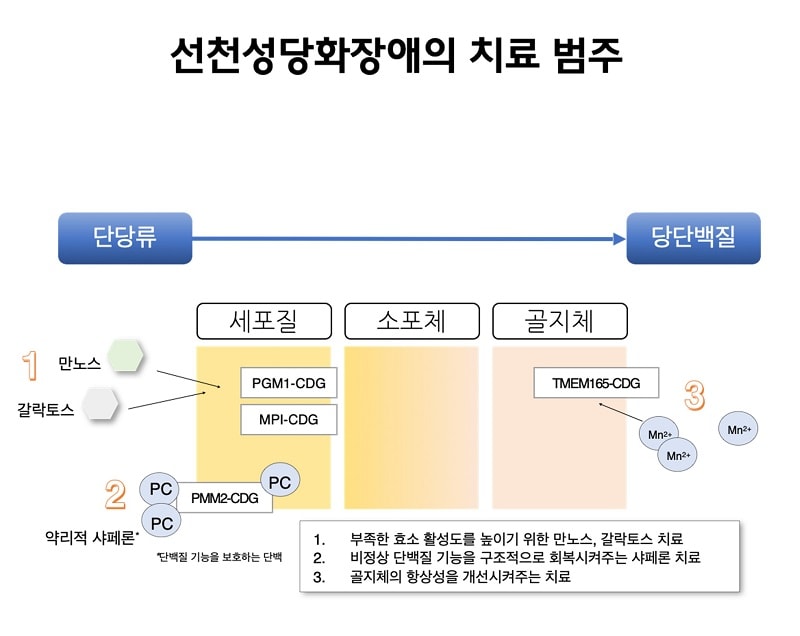
유전자의 이상에 의하여서 발생하는 질환이기 현재로서는 뚜렷한 근본적인 치료가 없는 실정이며, 특히 가장 많은 아형을 차지하고 있는 1a형은 많은 연구들에서 다양한 식이요법 등의 치료에 반응을 보이지 않았습니다. 1b형의 경우 만노스(mannose) 라는 당질의 보충이 도움이 된다고 알려져 있으며, 2c 형의 경우 과당(fructose) 보충이 도움이 된다고 알려져 있습니다. 아직까지 완치법이 없는 것은 사실이나 다양한 증상에 대한 보존적 치료를 통해 생존율 및 삶의 질의 향상을 기대할 수 있습니다. 발달지연 및 경련이 나타나는 경우에는 항경련제를 사용해 볼 수 있으며, 저혈당증에 대한 모니터링 및 발생 시 이에 대한 처치가 필요합니다. 일부 아형의 CDG의 환자는 간이식을 통해 증상이 개선되었다는 보고도 있습니다.
참고문헌
유한욱. 증례로 배우는 유전성 대사질환. 고려의학 2016년. P.688-692
Freeze HH, Eklund EA, Ng BG, et al. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012 May;11(5):453-66.
Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010 Dec;1214:190-8.
Grunewald S, Matthijs G, Jaeken J. Congenital disorders of glycosylation: a review. Pediatr Res. 2002 Nov;52(5):618-24.
Vaes L, Tiller GE, Pérez B, et al. PMM2-CDG caused by uniparental disomy: Case report and literature review. JIMD Reports. 2020;54:16–21.
Péanne R, de Lonlay P, Foulquier F, et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur J Med Genet. 2018 Nov;61(11):643-663.
Ferreira CR, Altassan R, Marques-Da-Silva D, et al. Recognizable phenotypes in CDG. J Inherit Metab Dis. 2018 May;41(3):541-553.
https://omim.org/entry/212065
Freeze HH, Eklund EA, Ng BG, et al. Neurology of inherited glycosylation disorders. Lancet Neurol. 2012 May;11(5):453-66.
Jaeken J. Congenital disorders of glycosylation. Ann N Y Acad Sci. 2010 Dec;1214:190-8.
Grunewald S, Matthijs G, Jaeken J. Congenital disorders of glycosylation: a review. Pediatr Res. 2002 Nov;52(5):618-24.
Vaes L, Tiller GE, Pérez B, et al. PMM2-CDG caused by uniparental disomy: Case report and literature review. JIMD Reports. 2020;54:16–21.
Péanne R, de Lonlay P, Foulquier F, et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur J Med Genet. 2018 Nov;61(11):643-663.
Ferreira CR, Altassan R, Marques-Da-Silva D, et al. Recognizable phenotypes in CDG. J Inherit Metab Dis. 2018 May;41(3):541-553.
https://omim.org/entry/212065
암 치료비 ∙ 보험금은
얼마일까?
얼마일까?
미리 계산하고
치료비 걱정을 덜어보세요.
치료비 걱정을 덜어보세요.
자세히


국내 유일,
중증 질환 맞춤형
보험 케어를 받고 싶다면?
중증 질환 맞춤형
보험 케어를 받고 싶다면?
내 치료 ∙ 마음 ∙ 재정 상태를
기반으로 케어해 드려요.
기반으로 케어해 드려요.
자세히


암 · 희귀질환과 관련된
전 세계 정보,
전 세계 정보,
레어노트에서 확인해 보세요
휴대폰으로 QR를 촬영하면
앱을 설치할 수 있습니다.

