

어깨고관절 점상 연골형성이상(1형-3형)
Rhizomelic chondrodysplasia punctata(type 1-3)
개요
어깨고관절 점상 연골형성이상은 과산화소체질환(peroxisomal disorder)* 중 하나로, 일차적으로 사지의 근위부(몸쪽 부분)에 영향을 미침에 따라 불균형적으로 작은 키가 특징입니다. 전형적인 얼굴의 모습은 콧등이 평평하고, 눈구석주름이 있으며, 입천장이 높고, 바깥귀(외이)의 기형과 작은턱증(소악증) 및 안구 이상이 동반됩니다. 선천성 구축, 왜소증, 심각한 정신 지체 및 경직을 보이기도 한다. 대부분의 환자는 생후 10년 내 사망하는 것으로 알려져 있습니다. 제1형부터 3형까지 세 가지 종류가 있으며, 이 중 제1형이 가장 흔합니다.
*과산화소체질환: 세포질 내 과산화소체의 기능 이상으로 인한 유전성 대사 이상 질환을 통칭함
원인
어깨고관절 점상 연골형성이상 제1형은 생화학적으로 플라즈마로겐(plasmalogen)의 생산과 피탄산(phytanic acid)의 알파산화에 결함으로 인하여 발생합니다. 제1형에서는 PEX7 유전자의 돌연변이로 인해 과산화소체생합성질환(peroxisomal biogenesis disorder, PBD)의 상보성집단 11(complementation group 11, CG11)에 해당하는 세포가 존재합니다. 제2형은 acyl-CoA: dihydroxyacetonephosphate acyltransferase 효소를 코딩하는 GNPAT 유전자(염색체 1q42 부위)의 돌연변이로 인하여 발생합니다. 제3형은 alkyldihydroxy- acetonephosphate synthase 효소를 코딩하는 AGPS 유전자(염색체 2q31 부위)의 돌연변이에 의합니다. 제1형은 여러 종류의 효소와 관계되므로 과산화소체생합성질환으로 분류되고, 제2형 및 3형은 단일 과산화소체효소 결핍증으로 분류됩니다.
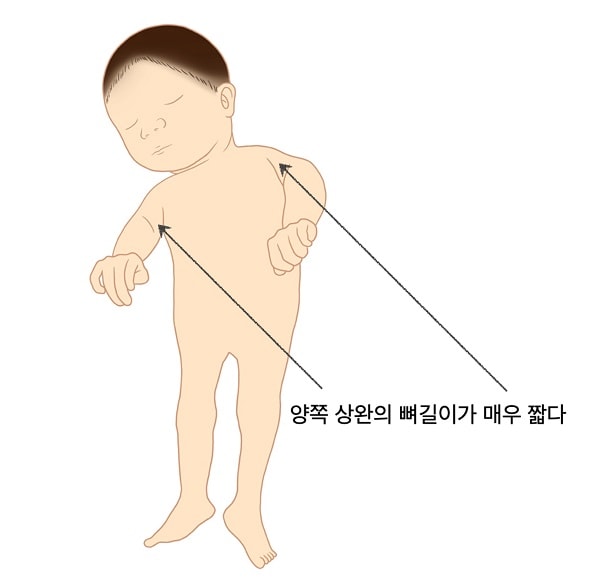
증상
환자는 위팔뼈의 몸 쪽 부분이 특히 짧고 대퇴골은 그보다 정도는 덜하지만 역시 짧습니다. 연골의 점상 석회화와 뼈끝(골단) 및 뼈몸통끝(골간단)의 이상 (점상연골형성장애) 척추골몸통의 관상 틈새(coronal clefts of the vertebral bodies), 및 백내장과 같은 증상이 태어날 때부터 또는 생애 첫 수개월 내 나타납니다.
출생 당시 체중, 키, 머리둘레는 정상범위보다 낮은 경우가 흔하며, 출생 후 성장지연도 두드러집니다. 심각한 인지기능 장애를 보이며, 대다수의 환자에서 발작이 나타납니다. 대부분의 환자는 생후 10년 내 사망하며, 상당수가 신생아기에 사망합니다. 일부 경한 증상을 보이는 환자도 선천성 백내장 및 연골형성이상을 보이는 것으로 알려졌으며, 어깨고관절 연골형성이상을 동반하지 않는 경우도 있고, 일부는 인지기능장애와 성장지연이 심하지 않은 경우도 있습니다.
진단
제1형의 진단은 임상 소견에 근거하여 이루어지며, 생화학적 검사 및 분자유전학적 검사에 의하여 확진 됩니다. 과산화소체 기능을 평가하는 생화학적 검사로는 적혈구 plasmalogen 농도, 혈장 피탄산 농도, 혈장 very long chain fatty acid의 농도가 있으며, 이 질환의 환자에서는 각각의 검사에서 (순서대로) 저하, 증가, 정상 농도를 보이는데, 과산화소체 기질 효소들의 수용체인 PEX7 단백질의 결함으로 인한 것입니다. 이 수용체를 코딩하는 PEX7 유전자의 돌연변이는 제1형의 원인이 됩니다.
제2형 및 3형의 진단 또한 임상 소견 및 생화학적 검사 소견에 의합니다. 제2형과 3형 모두 Plasmalogen 농도는 저하, 피탄산 농도는 정상입니다.
치료
보존적 치료를 행하나 출생 시부터 존재하는 다양한 장애에 의하여 한계가 있으며 예후가 좋지 않습니다. 백내장 적출로 인해 약간의 시력을 회복할 수 있고, 구축을 개선하기 위하여 물리치료가 추천됩니다. 일부에서 정형외과적 수술로 기능이 개선될 수 있습니다.
제1형의 경미한 증상의 환자에서는 피탄산의 축적으로 인한 증상을 예방하기 위하여 피탄산 제한 식이를 시행할 수 있습니다. 식사에 어려움이나 흡인이 반복되면 위루관 설치가 필요할 수 있습니다. 또한 호흡기능에 유의하여 인플루엔자 백신 및 RSV 단클론항체 주사를 고려하고, docosohexanoic acid(DHA)가 결핍된 환자에서는 보충요법을 시도 할 수도 있습니다. 성장 발달을 모니터하고, 발작 조절, 시력, 청력, 구축, 정형외과적 합병증에 대한 정기적인 평가를 시행합니다.
제1형은 상염색체 열성으로 유전되며, 환아의 부모에서 매 임신 시마다, 부모로부터 돌연변이 대립유전자를 모두 받아 환자가 될 확률은 25%이며, 50%의 확률로 보인자가 되고, 돌연변이 대립유전자가 없는 정상인이 태어날 확률은 25%입니다. 환자에서 돌연변이가 확인되면, 환자의 가족에서 잠재적인 위험이 있는 친척의 보인자 여부 확인 및 임신시의 산전검사를 위하여 분야유전학적 검사를 시행할 수 있습니다.
참고문헌
Shanske AL, Bernstein L, Herzog R. Chondrodysplasia punctata and maternal autoimmune disease: a new case and review of the literature. Pediatrics. 2007;120:e436-41.6.
Abdullah Çim, Salih Coşkun, et al. Rhizomelic Chondrodysplasia Punctata Type 1 Caused by a Novel Mutation in the PEX7 Gene. J Clin Res Pediatr Endocrinol. 2015;7(1):69-72.
https://www.ncbi.nlm.nih.gov/books/NBK1270/
http://www.omim.org/215100
http://www.omim.org/222765
http://www.omim.org/600121
https://www.orpha.net/consor/cgi-bin/Disease_Search_Simple.php?lng=EN&diseaseGroup=Rhizomelic+chondrodysplasia+punctata
https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=3567&Disease_Disease_Search_diseaseGroup=Rhizomelic-chondrodysplasia-punctata&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Rhizomelic-chondrodysplasia-punctata&title=Rhizomelic%20chondrodysplasia%20punctata&search=Disease_Search_Simple
얼마일까?
치료비 걱정을 덜어보세요.


중증 질환 맞춤형
보험 케어를 받고 싶다면?
기반으로 케어해 드려요.


전 세계 정보,

